


在科研学习中,经常会听到SNP这个概念,然而SNP究竟是什么呢,我们为什么要研究SNP,又如何开展研究工作呢?下面就一起来看一看吧。
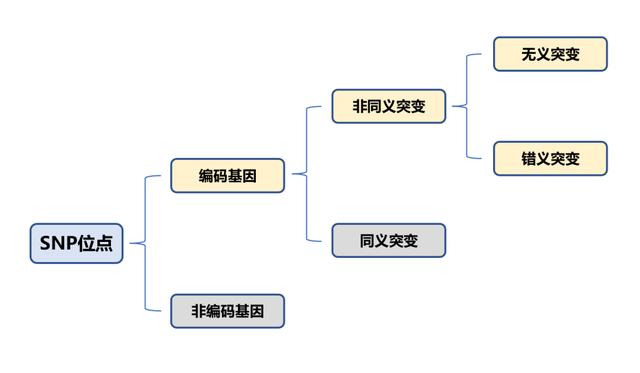
单核苷酸多态性(single nucleotide polymorphism , SNP),指由单个核苷酸发生转换、颠换、插入或缺失引起的DNA序列多样性,变异频率>1%。SNP广泛存在,人体内总共约有3×106个SNP位点,平均500-1000个碱基对中就会有1个。在人体数量庞大的SNP位点中,仅有少部分会引起氨基酸的变化,这取决于SNP的位置和突变种类。如下图所示,只有在编码基因区发生非同义突变的SNP才会引起表型的变化。
SNP与个体的表型差异、药物敏感、疾病易感性等相关,在精准营养、疾病诊断筛查和用药指导等方面具有重要的研究价值,与我们的生活息息相关。已有研究报道发现了与新冠肺炎症状具有潜在关联的SNP位点及相关基因[1],甚至新型冠状病毒,在进化演变过程中,也出现了很多SNP位点,其中的一些变异甚至使得新冠的传染性和毒力存在进一步增强的潜在风险。
针对SNP的研究可以分为2大类:
1、对未知SNP的分析,包括发现新的SNP位点和确定某一未知SNP和某种遗传疾病之间的关系;
2、对已知SNP的分析,包括对不同群体SNP的遗传多样性研究和遗传疾病的基因诊断。
今天我们就来看一下,检测 SNP常用的方法都有哪些。
对于SNP的检测大多是通过PCR和测序进行,通过检测可以确定突变的碱基和位点。根据检测通量可将检测方法分为低通量和高通量两大类,低通量方法一次实验可检测几个到几十个SNP,高通量方法一次性可以检测成千上万个SNP位点,成本也相应较高一些,在实际应用中可以根据实验需要选择合适的方法。首先我们来看一下低通量的方法,包括Sanger测序、Taqman探针和质谱仪检测等方法。
一、Sanger测序
Sanger测序是依赖于双脱氧核苷酸终止反应,产生不同长度片段进行测序的,是SNP检测的“金标准”,Sanger测序不仅可以确定突变的类型和位置,还能发现未知SNP位点。
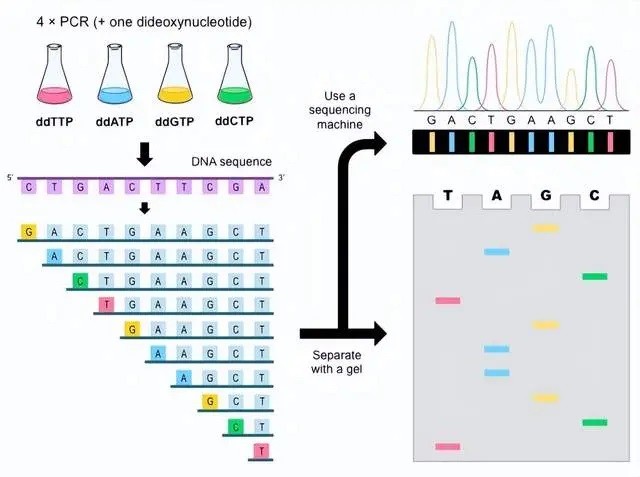
Sanger测序成本相对较高,但通量较低,适用于位点少,样本少的情况。
二、Taqman探针法
探针法是一项基于qPCR平台的,具有速度快、特异性好、灵敏度高、准确性高等特点的SNP分型技术。
探针5’端带有荧光基团,3’端带有淬灭基团,PCR过程中,探针与DNA模板结合,Taq酶延伸并水解探针,释放荧光,通过仪器实时监测荧光信号的强度。在SNP分型实验中,会根据不同的基因型设计含有对应碱基的探针,并带上不同的荧光基团用以区分,同时加上MGB基团使碱基错配的探针与模板结合的能力大打折扣,被剪切的概率也降低,因此被检测到的信号很弱,而能正确配对的探针则可与模板结合而被检测荧光信号,从而实现分型。
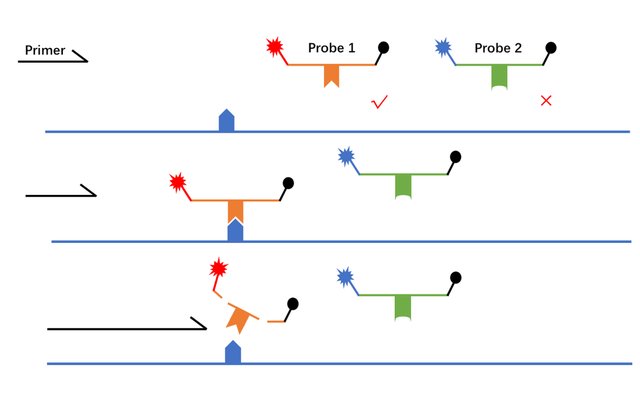
Taqman探针法成本较高,适用于少量SNP位点,大量样本的分析,因为单个位点探针设计昂贵,若样本量较大,则成本会降低。
三、质谱仪检测
质谱法是基于基质辅助激光解析电离飞行时间质谱技术(MALDI-TOF MS)的检测方法,其原理及操作如下:
(1) 通过PCR扩增含有SNP的目的片段(SNP位点前后约50 bp的片段);
(2) 使用虾碱性磷酸酶(SAP)去除体系中的底物及引物,加入单碱基延伸引物(3’末端与SNP位点前一碱基互补)和ddNTP;
(3) 得到3’末端ddNTP与SNP位点等位基因对应的产物片段,则产物片段仅比单碱基延伸引物多一个与SNP互补的ddNTP;
(4) 飞行质谱检测产物片段分子量,与单碱基延伸引物比较即可获得SNP位点的碱基信息。
该法第一步中PCR产物在进行单碱基延伸前需要先纯化,单碱基延伸产物在质谱上机前也需要纯化,操作较复杂,多用于医学领域。
上述几种SNP检测方法均属于低通量方法,当一次实验想获得大量SNP位点信息时,就需要用到高通量的方法,高通量方法多以二代测序技术为基础,主要包括:全基因组重测序、全外显子捕获测序、简化基因组测序和基因芯片等方法。
一、全基因组重测序
全基因组重测序(whole genome sequence, WGS)对已知基因组序列物种的不同个体进行全基因组测序,通过序列对比可以获得SNP位点信息。
WGS的实验原理如下:提取样本的DNA,将基因组DNA随机打断,电泳回收200-500 bp的片段,两端加上接头后构建文库,利用二代测序(以illumina平台为例)方法,通过桥式扩增制备Cluster,采用边合成边测序的方式获得全基因组序列。
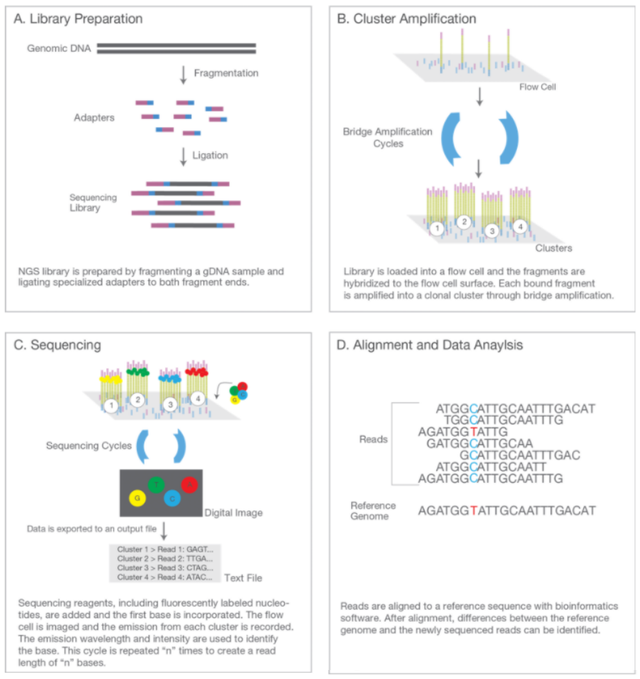
WGS通过生信手段分析个体基因组间结构的差异,除研究SNP外,还可以对结构变异位点(SV)、拷贝数变异位点(CNV)等进行检测。相对而言成本较高,并且对基因间区、内含子和外显子序列的SNP均进行检测,对特定目标区域的研究效率不高。
二、全外显子捕获测序
全外显子捕获测序(whole exome sequence, WES) ,WES是在WGS的基础上,仅针对外显子区域进行捕获测序,然后与参考基因组进行比较,筛选SNP位点的位置。WES实验主要包含文库构建、外显子靶向捕获和测序分析步骤,在实验中有一些问题需要注意。
(1)文库构建:将基因组DNA打碎成200-500 bp,或用转座酶处理成300 bp片段,然后构建DNA文库,在各片段两端加接头及index,进行片段筛选后扩增纯化;
(2) 外显子靶向捕获:用带有生物素的外显子探针库与文库进行杂交,然后探针与带有链霉亲和素的磁珠结合,将捕获外显子序列的磁珠收集并洗脱回收DNA,PCR扩增建库;
(3) 测序分析:与WGS一样,基于二代测序技术,对外显子文库基因序列进行测定。
在文库构建过程中,需要注意样本质量、index、接头浓度等因素对于文库质量影响较大,在实验起始时应保证DNA无严重降解,纯度较好并且起始量充足(gDNA≥50 ng,FEPP≥100 ng)。并且为避免后续测序过程中样本标签可能会发生错配,可以通过添加UDI/UMI接头减少错配概率,并在后续分析时将错配数据过滤。
相较于WGS,WES成本更低,性价比高,适用于大样本高深度研究,捕获测序区域明确,极大提高了特定目标区域的研究效率。
三、简化基因组测序
简化基因组测序是通过限制性核酸内切酶对基因组DNA进行酶切富集,酶切的片段在基因组上几乎随机分布,包含内含子、外显子及其他区域。同一个物种的酶切位点相对稳定,所以不同个体检测到的片段基本一致。简化基因组测序主要步骤为:
a. 对基因组进行酶切;
b. 挑选一定长度的酶切DNA片段回收建库测序
目前较常使用的有2b-RAD和Super GBS两种技术。
2b-RAD技术使用IIB型限制性核酸内切酶(如BsaXI)进行酶切,该种酶的切割位点位于识别位点的两侧,酶切产物为33 bp的标签序列,这些标签富集后可用于高通量测序,通过生信分析实现全基因组范围SNP高通量筛查和分型。
Super GBS的原理是使用甲基化敏感型限制性核酸内切酶将基因组DNA进行酶切,获得基因区域片段,然后对酶切片段进行磁珠纯化,选择长度相对均一的片段建库,通过高通量测序,分析获得SNP信息并进行基因分型,是一种快速、简便、低成本的基因分型方法。
当基因组较大,重复序列较多时,选择Super GBS技术,样本有降解时,优先选择2b-RAD技术。
四、基因芯片
基因芯片(gene chip),也是基于单碱基延伸原理的SNP检测方法。芯片的大小与玻片相仿,表面有微米级的凹槽,每个凹槽嵌入一个表面含有大量独立且密集寡核酸探针的微珠,探针的设计有两种,第一种探针的序列尾部最后一个碱基对应SNP位点前一个碱基,待测片段与微珠表面探针结合,ddNTP上被不同的荧光染料标记(A、T为绿色荧光,C、G为红色荧光),通过扫描检测芯片上的荧光信号确定SNP的类型,此时可以检测到A-C,A-G,T-C和T-G突变类型;第二种探针尾部最后一个碱基则是覆盖到SNP位点所在的碱基,若能够互补,则可以延伸,加上一个带染料的ddNTP,扫描有荧光;若不能互补,则不能延伸,扫描无荧光。
本文介绍了目前常用的SNP检测方法,每种方法都有其适用的场景和特点,在实际科研中,大家可以根据实验的具体需求选择合适的方法。